|
Extreme-Scale Visualization and Analysis of Fluid-Structure Interactions: HARVEY
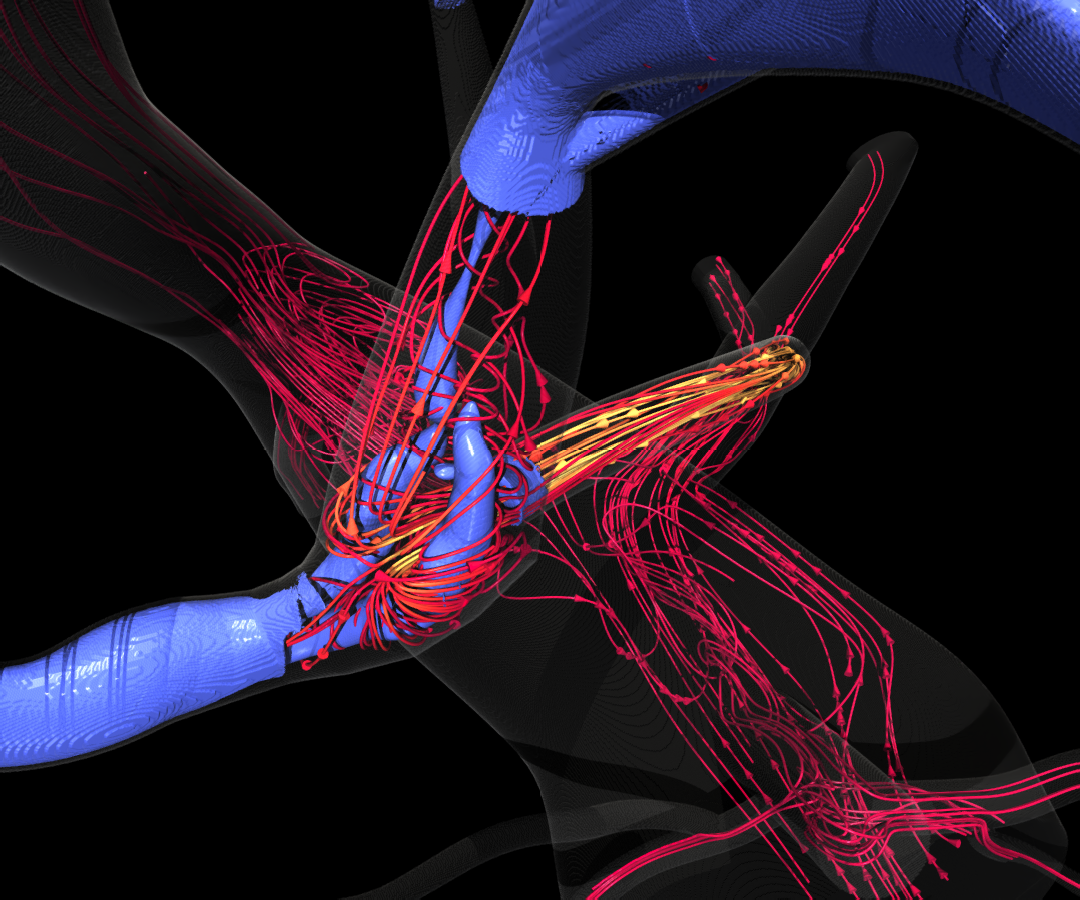
HARVEY, a massively parallel computational fluid dynamics code that predicts and simulates how blood cells flow through the human body, is used to study the mechanisms driving disease development, inform treatment planning, and improve clinical care. A Duke University-led team of researchers aims to repurpose HARVEY to improve our understanding of metastasis in cancer.
Challenge
One in four deaths in the United States is due to cancer, and metastasis is responsible for more than 90 percent of these deaths. The metastatic patterns of circulating tumor cells (CTCs) are strongly influenced by both a favorable microenvironment and mechanical factors such as blood flow. Advancing the use of data science to drive in-situ analysis of extreme-scale fluid-structure-interaction (FSI) simulations, this work aims to leverage the ALCF’s exascale Aurora system to model and analyze the movement of CTCs through the complex geometry of the human vasculature and thereby lay the groundwork for a predictive model of cancer metastasis. Simulating the rare cells, nearby red blood cells, and underlying fluid of the arterial network presents not only a computationally challenging simulation but a large data problem for posterior analysis. Scalable and in-situ analysis of massively parallel FSI models, including cellular-level flow, will be critical for enabling new scientific insights into the mechanisms driving cancer progression.
HARVEY is based on the lattice Boltzmann method (LBM) for fluid dynamics. Advantages of LBM over other numerical solvers of the Navier-Stokes equations include its amenability to parallelization due to its underlying stencil structure and the local availability of physical quantities, eliminating the need for global communication among processors required of Poisson solvers.
HARVEY adopts for execution on GPUs the MPI+X parallelization model, where X was originally OpenMP for CPUs and CUDA. The code has easily separable kernels that are optimized uniquely for each system’s architecture to take advantage of system features and avoid potential performance pitfalls for the computation of three primary components: task distribution, density and momentum vector, and collision and equilibrium relaxation. Over the last few years HARVEY has been ported to SYCL, HIP, and Kokkos to enable functionality and performance on a variety of supercomputing systems, each with different hardware.
Performance Results
As of 2024, the researchers have demonstrated HARVEY’s functionality on Aurora for two distinct cases. The first is for fluid-only calculations and the second for fluid and red-blood cell simulations. In both cases, HARVEY ran on full-body human vasculatures. The researchers successfully performed fluid-only simulations on as many 2048 Aurora nodes. The team deployed HARVEY on 1024 nodes for red-blood cell and fluid simulations.
The researchers have successfully built the code’s framework, on Aurora, on top of which in the Kokkos version they integrated in-situ visualization. This integration has been verified by rendering the fluid velocity field generated by the in-situ visualization of a ”fluid in a tube” test case.
Impact
The exascale-optimized HARVEY application will offer the ability to create personalized models for individual patients. Blood flow simulations have the potential to greatly benefit diagnosis and treatment of patients suffering from vascular disease. By predicting where metastasized cells might travel in the body, HARVEY can help doctors anticipate early on where secondary tumors may form. Empowering models of the full arterial tree can provide insight into diseases such as arterial hypertension and enable the study of how local factors impact global hemodynamics.